Target-Based Drug Design: Role ofMolecular Docking in Anti-Cancer DrugDiscovery
DOI:
https://doi.org/10.64062/JPGMB.Vol1.Issue1.5Keywords:
- Molecular Docking, Target-Based Drug Design, Anti-Cancer Drug Discovery, Kinase Inhibitors, Virtual Screening, Computational Drug Design, Autodock, Binding Affinity
Abstract
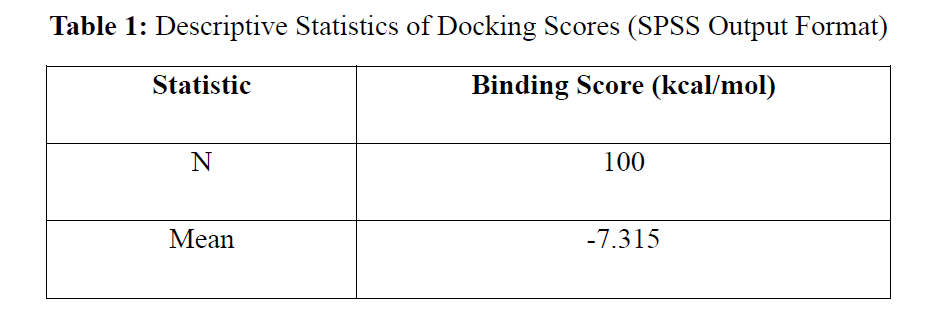
The study explores a drug design targeting HER2 receptor as a leading target in multiple cancers, is presented based on molecular docking simulations to find novel anticancer drugs. With the optimal structures of proteins and ligands obtained through AutoDockTools and Open Babel, a library of 100 ligands was docked by AutoDockVina. The top 10 ligands exhibited strong binding affinities (−9.6 to −8.5 kcal/mol) and formed stable hydrogen bonds and hydrophobic interactions with the key active site residues on HER2. Hierarchical clustering was applied by R software to classify compounds with the same binding mode. In comparison with the work available in literature, the current study brings in a robust computational pipeline which combines molecular docking and statistical clustering with a particular emphasis on HER2. Despite being an in silico analysis, the study emphasizes that molecular docking could be a promising metric to exploit in early stage of anti-cancer drug discovery and suggests the need for wet-lab validation via molecular dynamics simulation, ADMET profiling and experimental studies.
References